

Einführung
Die Familiäre Hypercholesterinämie ist einer der häufigsten angeborenen Stoffwechselstörungen. Bei der Erkrankung können bereits in jungen Jahren Schäden an den Gefäßen entstehen, welche vom Patienten zunächst nicht bemerkt werden, im weiteren Verlauf jedoch zu einem deutlich erhöhten Risiko für Herz-Kreislauferkrankungen führen. Wird die Erkrankung erkannt, kann sie gut therapiert werden. Oft erfolgt die Diagnosestellung jedoch erst dann, wenn bereits deutliche Gefäßschäden vorliegen oder gar ein schweres Ereignis, wie ein Herzinfarkt oder Schlaganfall aufgetreten sind. Ziel von VRONI und VRONIPLUS ist eine frühzeitige Diagnostik und Therapie betroffener Patienten, damit Gefäßschäden möglichst erst gar nicht entstehen und der Langzeitverlauf somit günstig beeinflusst wird.
Familiäre Hypercholesterinämie (FH) – Was ist das?
| Familiär | bedeutet: von einem oder beiden Elternteilen vererbt. |
| Hyper | drückt aus: „zu hoch“. |
| Cholesterin | ist ein Fett (Lipid) und ein Naturstoff des menschlichen Körpers. Hier ist der Anteil des LDL-Cholesterins im Blutplasma gemeint. |
| Ämie | bezeichnet: das Vorkommen im Blut. |
FAMILIÄRE HYPERCHOLESTERINÄMIE (FH)
bedeutet also, dass die Erkrankung von den Eltern vererbt wird und mit einem „zu viel“ an LDL-Cholesterin im Blut einhergeht.

Stammbaum – Wer ist von FH betroffen?
Unsere Erbinformation (= DNA – Gesamtheit aller Gene in Form von Chromosomen), d. h. der Bauplan eines jeden Menschen liegt immer in doppelter Ausführung vor: Ein Gen stammt jeweils von der Mutter, das andere vom Vater.
Die Vererbung der FH erfolgt nach dem autosomal dominanten Erbgang. Das bedeutet, die Erkrankung tritt bereits auf, wenn nur ein defektes Gen (d. h. das von Vater oder Mutter) vorliegt.
Wenn ihr Kind an FH erkrankt ist, sind also möglicherweise auch Sie von FH betroffen. Dann stellt sich die Frage: Von wem haben Sie FH geerbt? Und von wem hat Ihr Kind die FH geerbt? Versuchen Sie zusammen mit Ihrem Kind den Stammbaum auszufüllen und so ein genaueres Verständnis für die Vererbung zu bekommen.
Wie häufig kommt FH vor?
In Deutschland gibt es keine systematische Datenerhebung zum Lipidstoffwechsel. Es kann nur geschätzt werden, dass ca. 162.000 bis 400.000 Menschen von FH betroffen sind. Weltweit sind es ca. 14 bis 34 Millionen.
Gut zu wissen: Was ist eigentlich der menschliche Stoffwechsel?
Stoffwechsel bedeutet so viel wie Austausch von Stoffen. Im weiteren Sinne heißt das: Stoffe, die wir über die Nahrung aufnehmen, werden durch die Darmwand in den Körper transportiert und vor allem von der Leber „verarbeitet“. Von dort aus werden Zucker, Fette und Eiweiße (aber auch andere Stoffe, wie z. B. Vitamine) im Körper verteilt: das Blut transportiert sie an die benötigten Stellen im Körper. Fette beispielsweise dienen dem Körper als Energieträger und -speicher. Eine Stoffwechselstörung bedeutet entweder, dass Nährstoffe vom Körper nicht abgebaut werden können oder aber nicht an den Orten ankommen, an denen sie benötigt werden.

Was ist Cholesterin?
Cholesterin ist ein fettähnlicher Stoff, der im Blut zirkuliert und vom Körper benötigt wird, um Zellen aufzubauen, Hormone zu produzieren und Gallensäure in der Leber zu erzeugen. Es kann sowohl vom Körper selbst produziert werden (von allen Organen, vor allem der Leber), als auch durch die Nahrung in Form von Fetten aufgenommen werden. Cholesterin besteht aus 2 Derivaten (d. h. „Abkömmlingen“): dem High Density Lipoprotein (HDL) und dem Low Density Lipoprotein (LDL). Lipoproteine wiederum sind kleine Pakete aus Cholesterin, Triglyceriden und Proteinen, die als Transportmittel für Fette im Blut dienen. So bringt LDL Cholesterin zu den Zellen, kann also als eine Art „Lieferservice“ betrachtet werden. Die Kinder lernen es als „Lass Das Lieber“-Cholesterin kennen, da es als die „ungesunde“ Form des Cholesterins gilt. HDL dagegen sammelt Cholesterin im Körper ein und bringt es zur Leber, wo es abgebaut wird. Es ist sozusagen die „Müllabfuhr“. Durch den Abtransport von Stoffen wird es als „gutes“ Cholesterin angesehen und gegenüber den Kindern als „Hab Dich Lieb“-Cholesterin bezeichnet. Neben HDL und LDL als Cholesterinderivate zählen außerdem noch Triglyzeride zu den Blutfetten. Sie dienen vor allem der Speicherung von Fett im Körper, um bei länger andauernder körperlicher Leistung Energie zu liefern.
ALLGEMEINER AUFBAU EINES LIPOPROTEINS [ Transportform des Cholesterins ]
Wie sind die Richtwerte für Cholesterin bei Kindern und Jugendlichen?
Nach den aktuellen Richtlinien der Arbeitsgemeinschaft für Pädiatrische Stoffwechselstörungen (Chourdakis, 2016) gelten bei Kindern und Jugendlichen LDL-Cholesterinwerte bis 110 mg/dl als noch akzeptabel, ab 130 mg/dl als erhöht. Bei HDL-Cholesterin wird eine Konzentration über 45mg/dl bei Kindern und Jugendlichen angestrebt.
MERKE
Zu hohe HDL-Werte gibt es in diesem Sinne nicht,
da viel HDL-Cholesterin vor fetthaltigen Ablagerungen an den Gefäßwänden schützt.
DIE „ATHEROSKLEROSE“
Wie sind die Richtwerte für Cholesterin bei Erwachsenen?
Die aktuell geltenden Richtlinien zu Blutfettwerten finden sich unter anderem bei der European Society of Cardiology (Mach et al., 2020). Hier kann ein Risiko-Score errechnet werden, der das Risiko für eine Herz-Kreislauf-Erkrankung abschätzt. LDL-Cholesterin sollte bei Erwachsenen unter 116 mg/dl (3 mmol/l) liegen, HDL-Cholesterin bei Frauen über 45 mg/dl (1,2 mmol/l) und bei Männern über 40 mg/dl (1 mmol/l). Diese Richtwerte gelten jedoch nur, wenn zusätzlich kein anderer Risikofaktor besteht – wie z. B. höheres Alter, männliches Geschlecht, Rauchen sowie das Vorliegen von Diabetes mellitus oder einer genetischen Belastung (z. B. FH). Bei erwachsenen Personen mit FH liegt der angestrebte Zielwert für LDL-Cholesterin bei unter 70 mg/dl.
Was genau passiert bei der FH?
Wie oben bereits erläutert, handelt es sich um eine erbliche Erkrankung, d. h. dass ein bestimmter Teil der Erbinformation (ein bestimmtes Gen) verändert (mutiert) ist. Die Mutationen betreffen den LDL-Rezeptor (häufigste Mutation), das ApoB100 (Lipoprotein) oder das PCSK9 Enzym.
LDL-Rezeptor
Ist für die Aufnahme von LDL aus der Blutbahn in die Zelle notwendig.
Apolipoprotein B100 [ kurz ApoB100 ]
ApoB100 ist ein Lipoprotein und vermittelt als „Verbindungsstelle“ die Bindung von LDL an den LDL-Rezeptor. Hierdurch gelangt das LDL in die Zelle. In der Zelle löst sich das LDL von seinen Rezeptoren und wird aufgespalten, während die Rezeptoren wieder aus der Zelle transportiert werden („Recycling“).
PCSK9
Dieses Enzym sorgt dafür, dass die mit LDL beladenen LDL-Rezeptoren abgebaut werden, ist also Teil der „Müllabfuhr“. Eine Veränderung in einem dieser 3 Gene führt dazu, dass das LDL-Transportsystem nicht richtig funktioniert. Das LDL-Cholesterin kann somit nicht oder nicht ausreichend vom Blut in die Zelle transportiert werden. Das LDL-Cholesterin kann somit in der Zelle nicht abgebaut werden und der Spiegel im Blut steigt an.
Was passiert, wenn der Cholesterin Spiegel dauerhaft erhöht ist?
Das LDL-Cholesterin kann sich an Wänden von Blutgefäßen festsetzen und so zu Gefäßverengungen und einem schlechten Blutfluss bis hin zu vollständigen Gefäßverschlüssen führen (Atherosklerose). Im weiteren Verlauf können kardiovaskuläre Erkrankungen, wie z. B. eine Koronare Herzkrankheit, ein Herzinfarkt oder ein Schlaganfall auftreten.
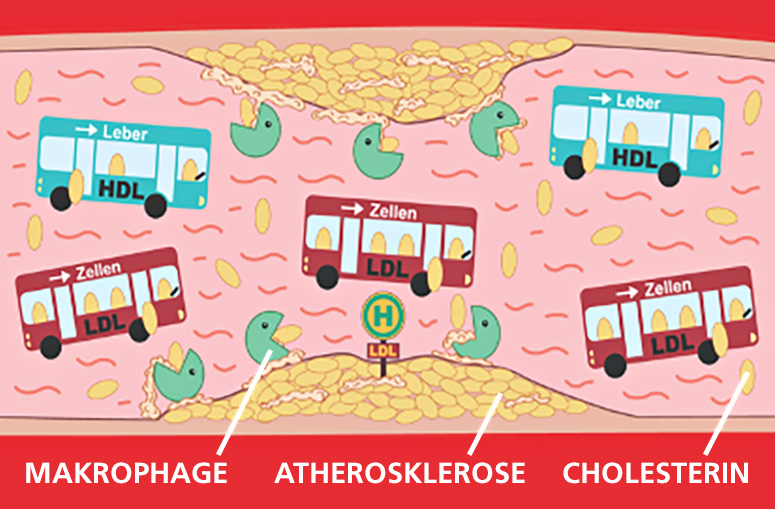
Bei der FH funktioniert der LDL-Transport (rote Busse) nicht, das LDL-Cholesterin verbleibt im Blut und setzt sich an den Gefäßwänden ab. Der HDL-Transport (mintfarbige Busse) sammelt das HDL ein und bringt es zur Leber, wo es abgebaut wird.
Welche Faktoren begünstigen die Entstehung von Gefäßschäden?
Neben der FH gibt es verschiedene weitere Einflussfaktoren, welche eine Atherosklerose begünstigen.
Zu diesen Risikofaktoren zählen:
Wie kann LDL-Cholesterin gesenkt werden?
1. Schritt
Lebensstil-Anpassung
[ Ernährung und Bewegung ]
2. Schritt
Medikamentöse Therapie
[ Monotherapie oder Kombinationstherapie ]
In den meisten Fällen ist eine Kombination aus 1. und 2. Schritt erforderlich, um den LDL-Spiegel ausreichend zu senken.
Ziel einer LDL-senkenden Therapie in der Kindheit und im Jugendalter
= den Cholesterinspiegel unter < 130mg/dl zu senken!
Ab dem achten Lebensjahr sollte eine medikamentöse Therapie in Erwägung gezogen werden, wenn die konsequente Lebensstilmodifikation und Ernährungstherapie über mehrere Monate keine befriedigende Veränderung der Cholesterinwerte erbringen.
Medikamente
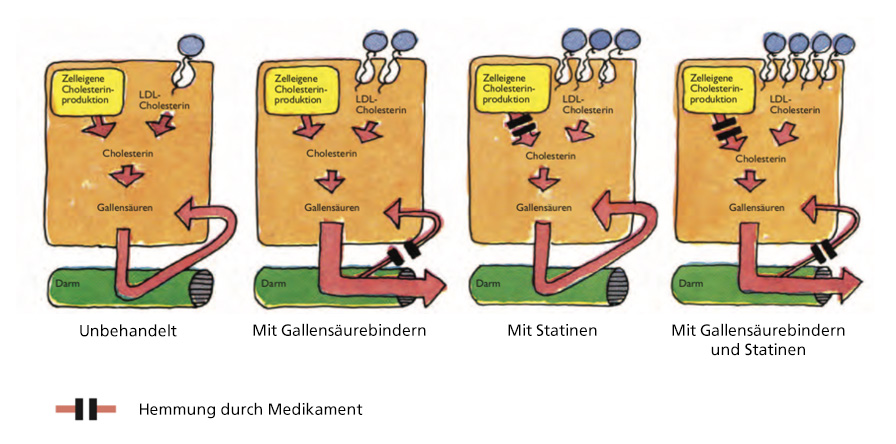
Therapie der ersten Wahl sind bei Kindern sogenannte Statine. Diese hemmen ein Enzym, das Cholesterin im Körper herstellt (HMG-CoA-Reduktase). Durch die Blockade des Enzyms wird in der Zelle ein Cholesterinmangel erzeugt, wodurch die Zelle mehr LDL-Cholesterin aus dem Blut aufnehmen muss. Hierdurch sinkt der LDL-Spiegel im Blut um bis zu maximal 50 % und das HDL-Cholesterin nimmt zu. Außerdem hemmen Statine Entzündungsprozesse in arteriosklerotischen Ablagerungen in den Gefäßwänden, sodass sich Ablagerungen nicht mehr so einfach bilden können. Bekanntes Präparat ist Pravastatin (ab 8 Jahren). Statine sind in der Regel gut verträglich.
Daneben kommen sogenannte Ezetimibe zum Einsatz, welche die intestinale Cholesterinaufnahme über den Darm verhindern. Sie sind ab dem 10. Lebensjahr zugelassen und weisen ebenfalls eine insgesamt gute Verträglichkeit auf.
| Chourdakis, M. B., S; Dokoupil, K; Oberhoffer, R; Schwab, KO; Wolf, M; Zimmer, KP; Koletzko, B. (2016). S2k-Leitlinien zur Diagnostik und Therapie von Hyperlipidämien bei Kindern und Jugendlichen. Arbeitsgemeinschaft für Pädiatrische Stoffwechselstörungen (APS) in der Deutschen Gesellschaft für Kinderheilkunde und Jugendmedizin, Berlin. |
| Hoc, S. (2004). Lipidstoffwechselstörungen: Wenn die Cholesterinwerte schon bei Kindern erhöht sind. Dtsch Arztebl International, 101(17), A-1184. Retrieved from https://www.aerzteblatt.de/int/article.asp?id=41600 |
| Klose, G., Laufs, U., März, W., & Windler, E. (2014). Familiäre Hypercholesterinämie. Dtsch Arztebl International, 111(31-32), 523-529. Retrieved from https://www.aerzteblatt.de/int/article.asp?id=161185 |
| Mach, F., Baigent, C., Catapano, A. L., Koskinas, K. C., Casula, M., Badimon, L., . . . Wiklund, O. (2020). 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J, 41(1), 111-188. doi:10.1093/eurheartj/ehz455 |
| Nordestgaard, B. G., Chapman, M. J., Humphries, S. E., Ginsberg, H. N., Masana, L., Descamps, O. S., . . . Tybjaerg-Hansen, A. (2013). Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J, 34(45), 3478-3490a. doi:10.1093/eurheartj/eht273 |
| Vuorio, A., Kuoppala, J., Kovanen, P. T., Humphries, S. E., Tonstad, S., Wiegman, A., . . . Ramaswami, U. (2019). Statins for children with familial hypercholesterolemia. Cochrane Database of Systematic Reviews(11). doi:10.1002/14651858.CD006401.pub5 |
VRONIPLUS Schulungszentrum
Lehrstuhl für Sozialpädiatrie der TU München · Lehrstuhl für Präventive Pädiatrie der TU München
Heiglhofstraße 65 · 81377 München · Telefon: 089 71009 392